

We are excited about some new work out of lab that relaxes an important assumption in phylogenetics. In a new bioRxiv preprint, Perry Wood, Jr., Cam Siler, Rafe Brown, and I describe how we generalize the space of trees in Bayesian phylogenetics to avoid assuming divergences are independent across a phylogeny, and allow us to infer shared divergences among multiple clades. We have released a new version of ecoevolity (Version 1.0.0) that implements this fully phylogenetic approach to inferring shared divergence events across phylogenetic trees.
The assumption
In phylogenetics, we assume lineages diverge independently (conditional on the topology) and only leave two descendant lineages (i.e., bifurcate). Put another way, we assume all processes of diversification cause only a single lineage to bifurcate, and have no affect on any of the other extant lineages across the phylogeny. Across the tree of life, there are likely many exceptions to this assumption. For example, we can imagine rising sea levels fragmenting an island and causing co-distributed species of lizards to diverge at the same time.
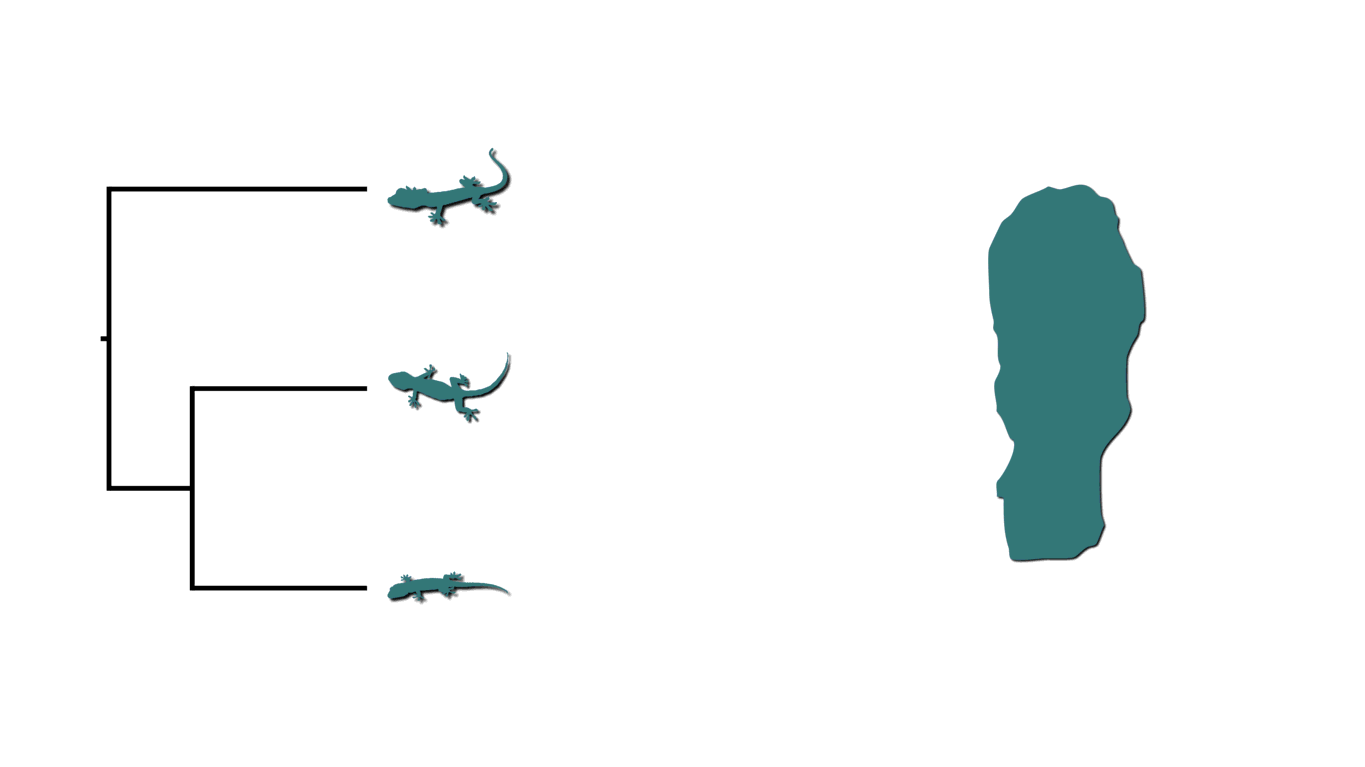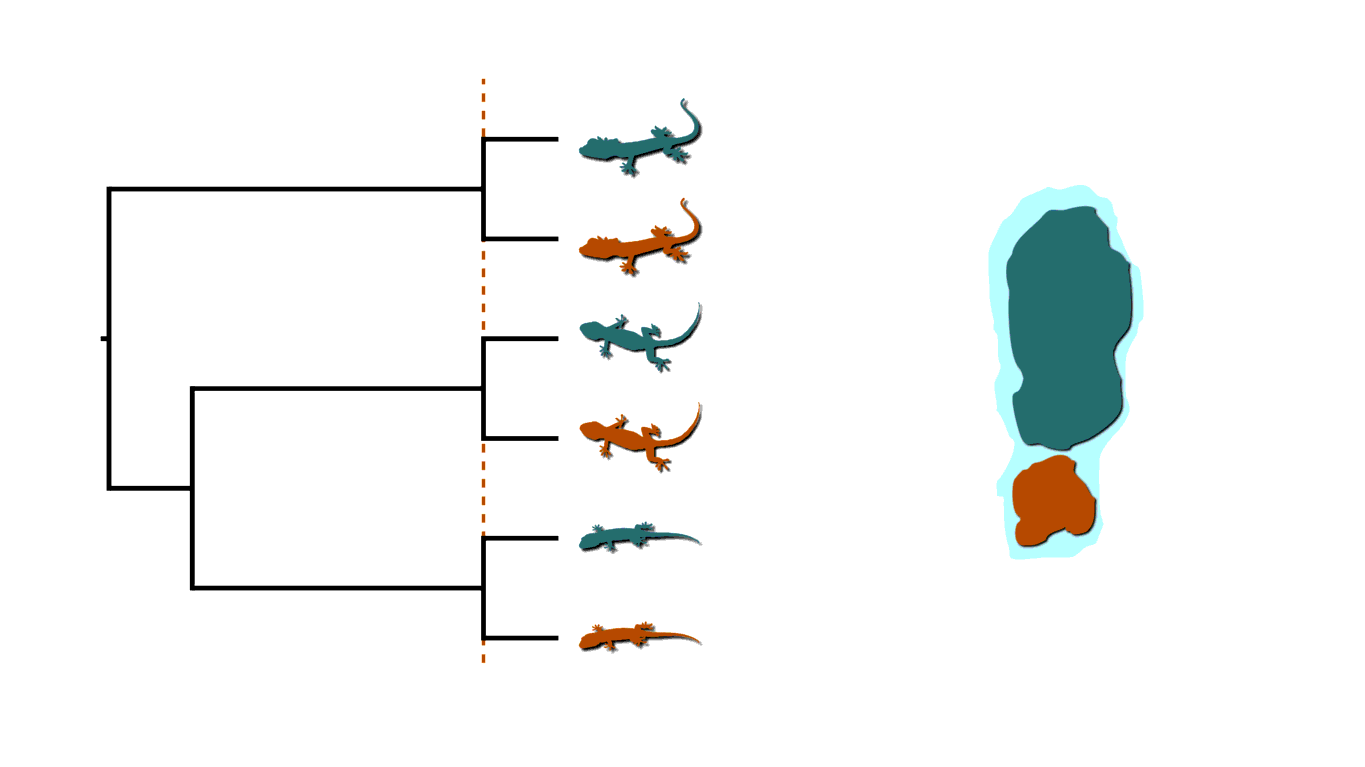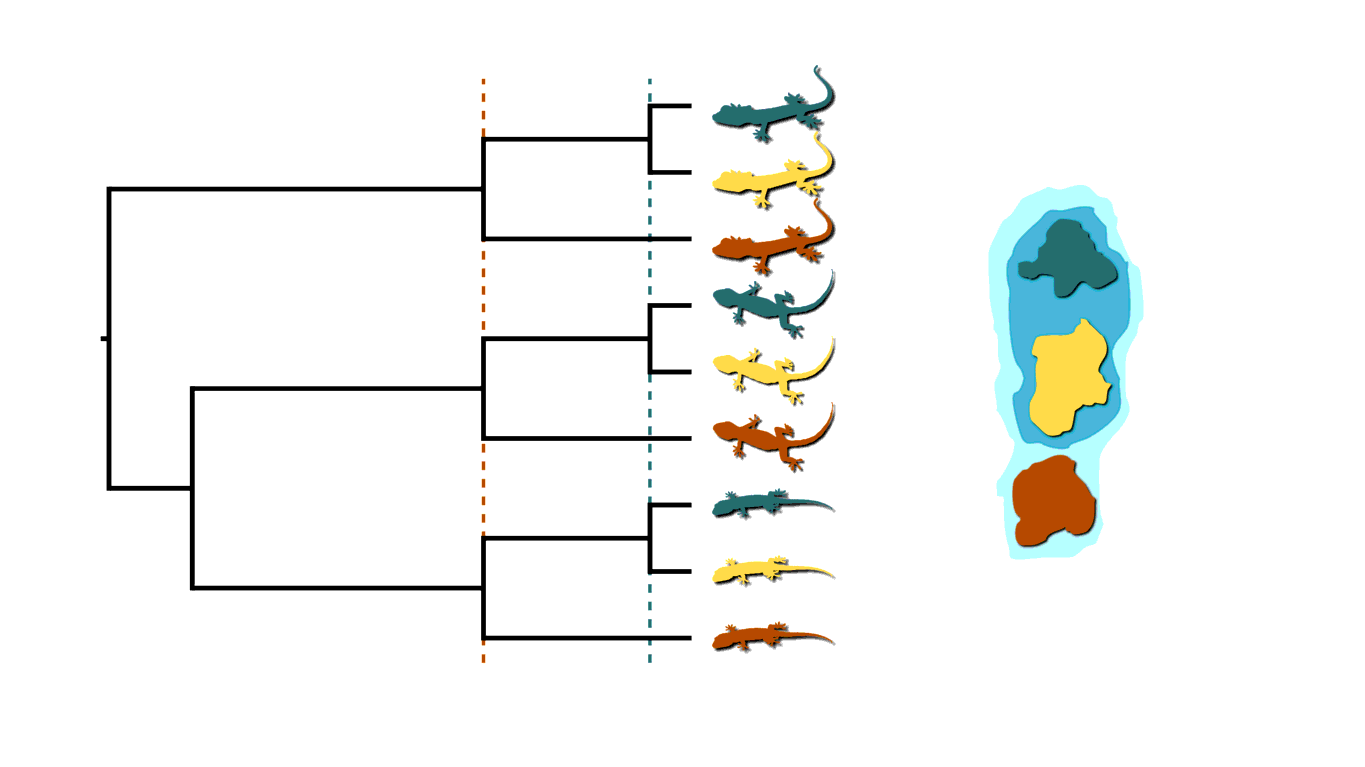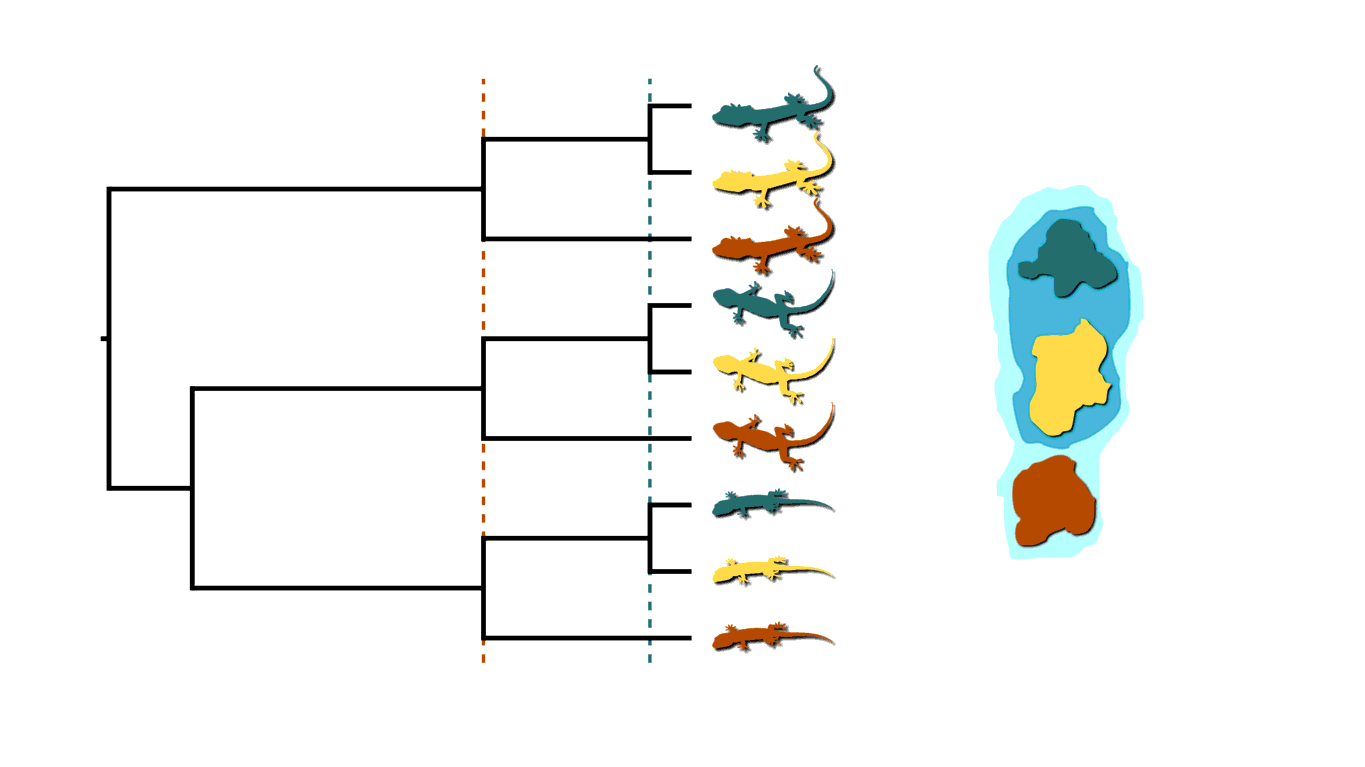
Island fragmenetaion causing bouts of shared divergences.
Instead of lizards on islands, let’s imagine three members of a gene family residing along a region of a chromosome that gets duplicated. This would create shared divergences across the phylogenetic history of the gene family. When multiple infected individuals spread a pathogen to others at a social gathering, this will create shared divergences in the “transmission tree” of the pathogen. In fields as diverse as biogeography, genome evolution, and epidemiology, there are interesting processes that violate the assumption of independent divergences and cause shared divergences.
Back to our insular lizard example, we can also imagine the rising sea levels fragmenting the island into more than two islands, like in the animation below. This will cause multifurcations (a lineage diverging into three or more descendants) in addition to shared divergences.
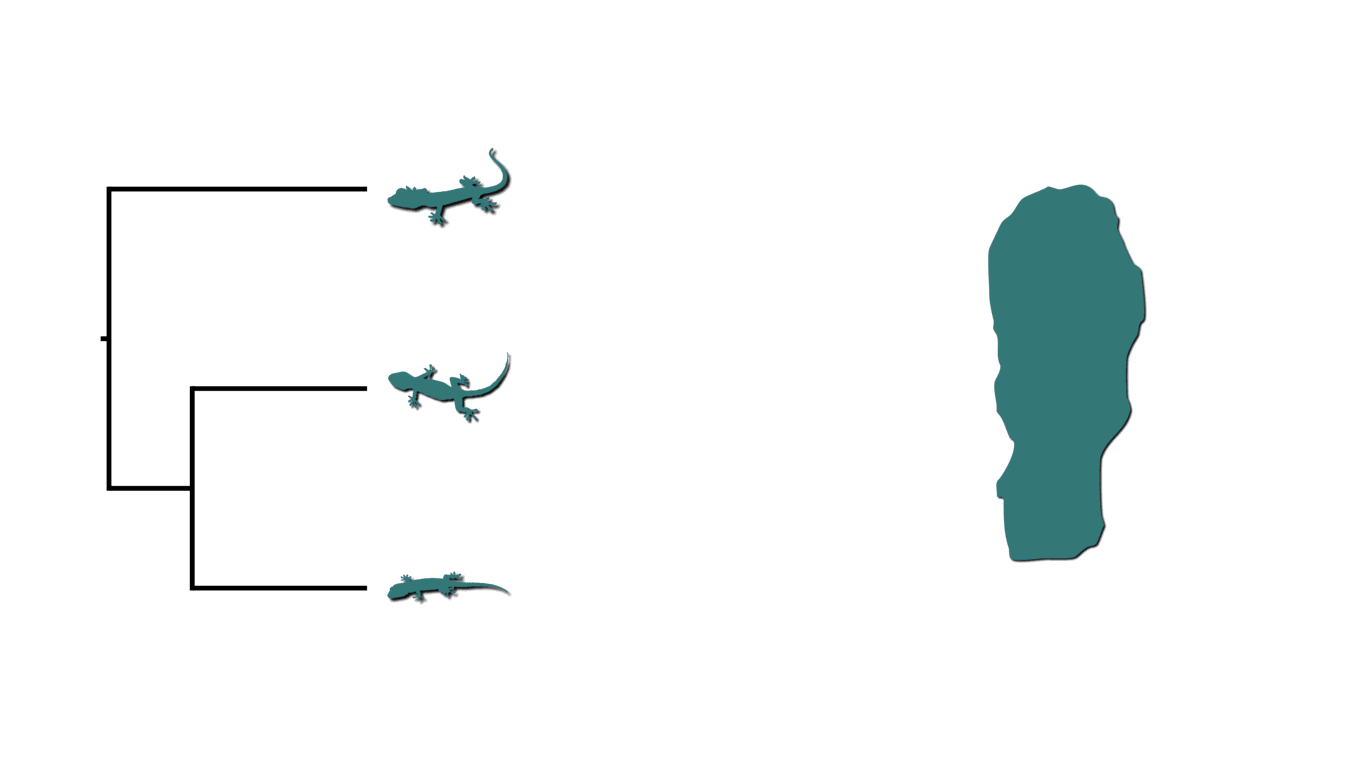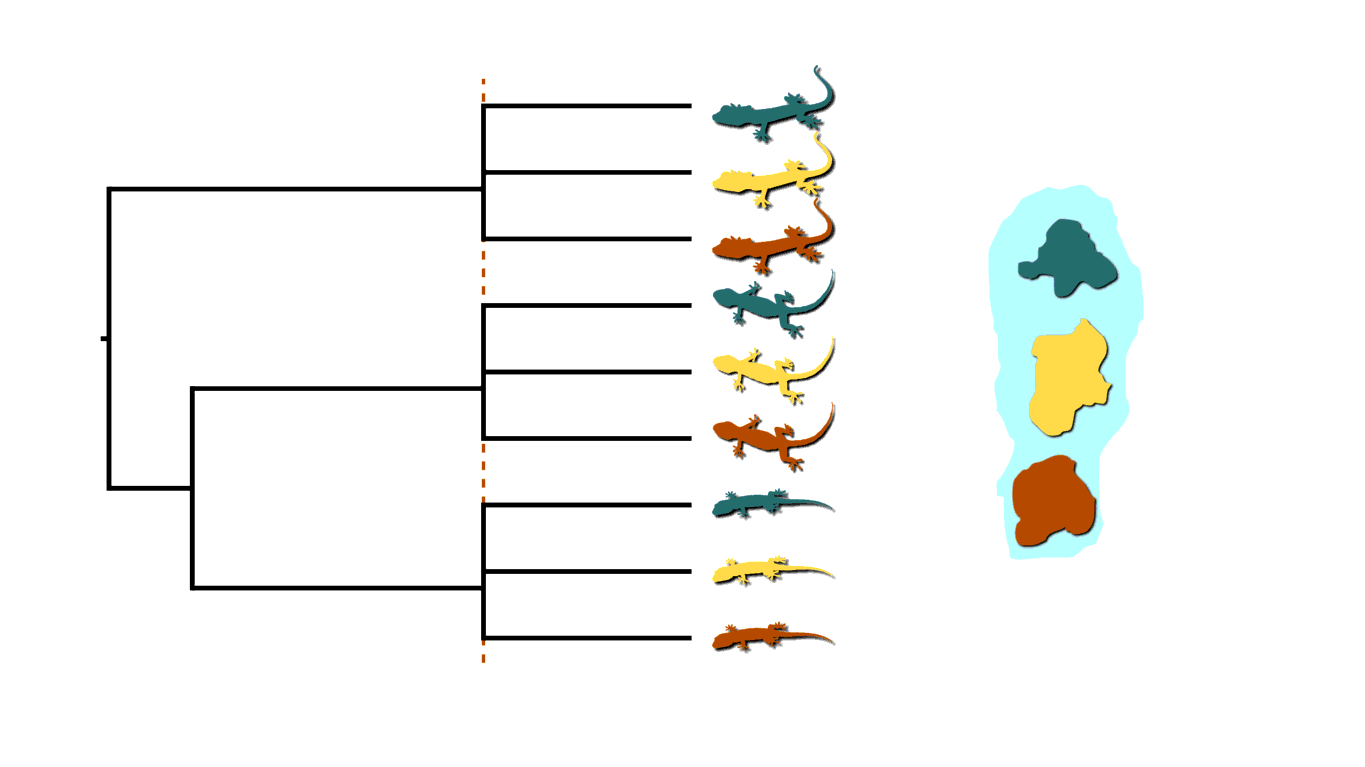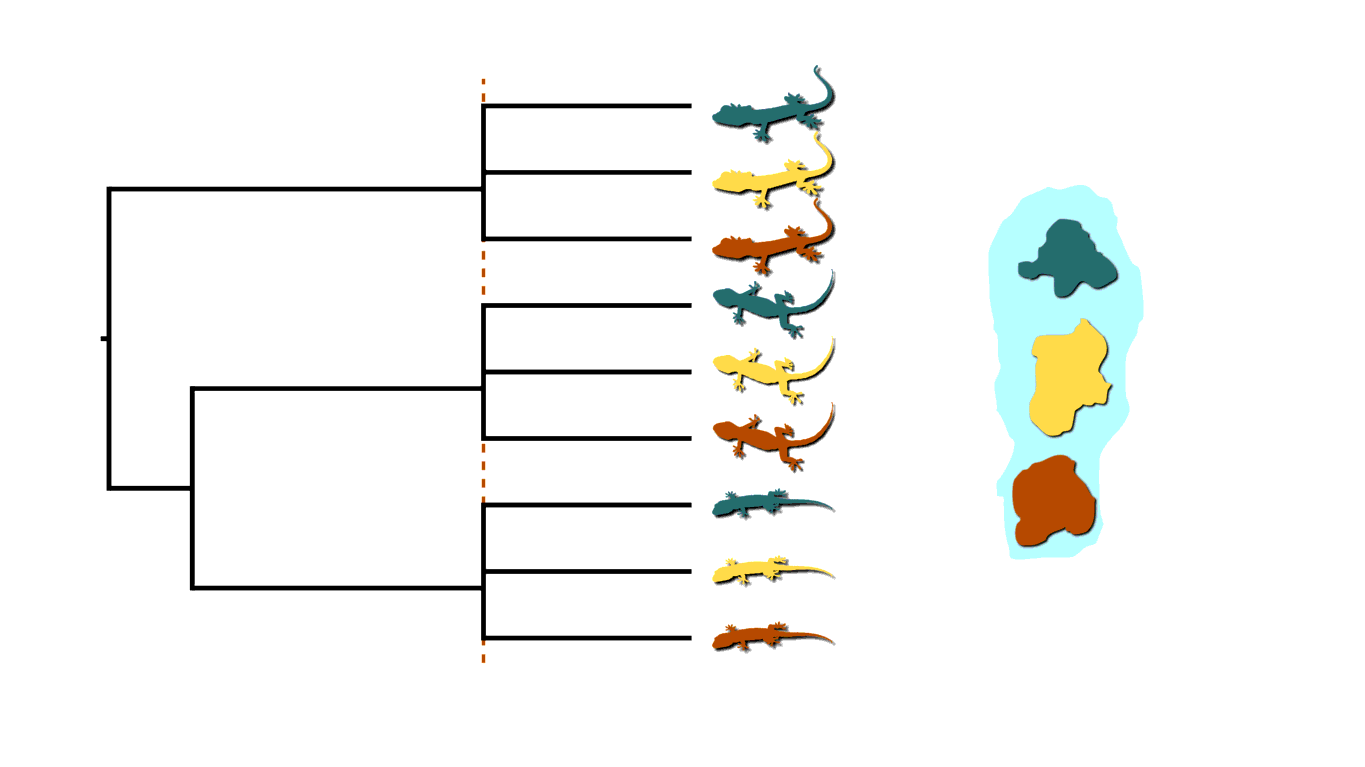
Island fragmenetaion causing multifurcating, shared divergences.
Similarly, in epidemiology, when an infected individual spreads a pathogen to two or more others at a social gathering, this will create a multifurcating divergence in the transmission tree.
Current phylogenetic methods for inferring rooted trees assume all divergences are independent and bifurcating. More formally, if we have \(N\) tips, current methods only consider trees with \(N-1\) independent, bifurcating divergences. When shared or multifurcating divergences were common in the evolutionary history of the system we want to study, such tree models are over-parameterized as illustrated below.

When shared or multifurcating divergences have occurred, current phylogenetic models are over-parameterized.
More importantly, by assuming all divergences are independent and bifurcating, current phylogenetic methods do not allow us to test for patterns of shared or multifurcating divergences predicted by processes of diversification that are of interest across fields like biogeography, genome evolution, and epidemiology.
Our approach
To relax this assumption, we developed a Bayesian approach to generalizing the space of tree models to allow for shared and multifurcating divergences (Oaks et al., 2022). Under our generalized tree distribution, trees with \(N - 1\) bifurcating divergences are only one class of tree models in a greater space of trees with anywhere from from 1 to \(N - 1\) potentially shared or multifurcating divergences.
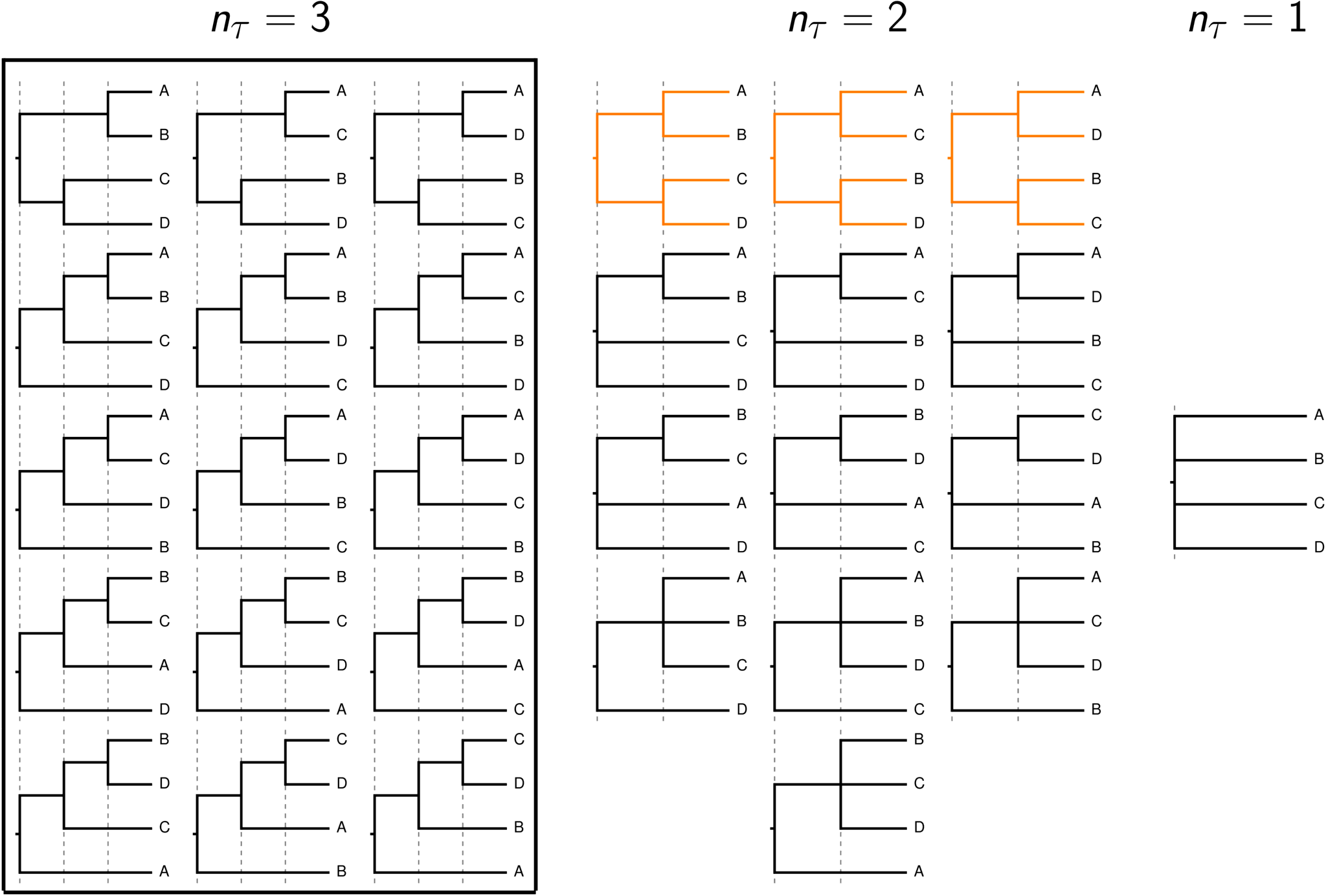
The rooted topologies considered by current phylogenetic methods (within box) and the additional tree models considered by phycoeval's generalized tree model. The tree models with nonindependent (shared) divergences are highlighted in orange.
We developed reversible-jump Markov chain Monte Carlo algorithms to perform inference across this generalized space of trees.
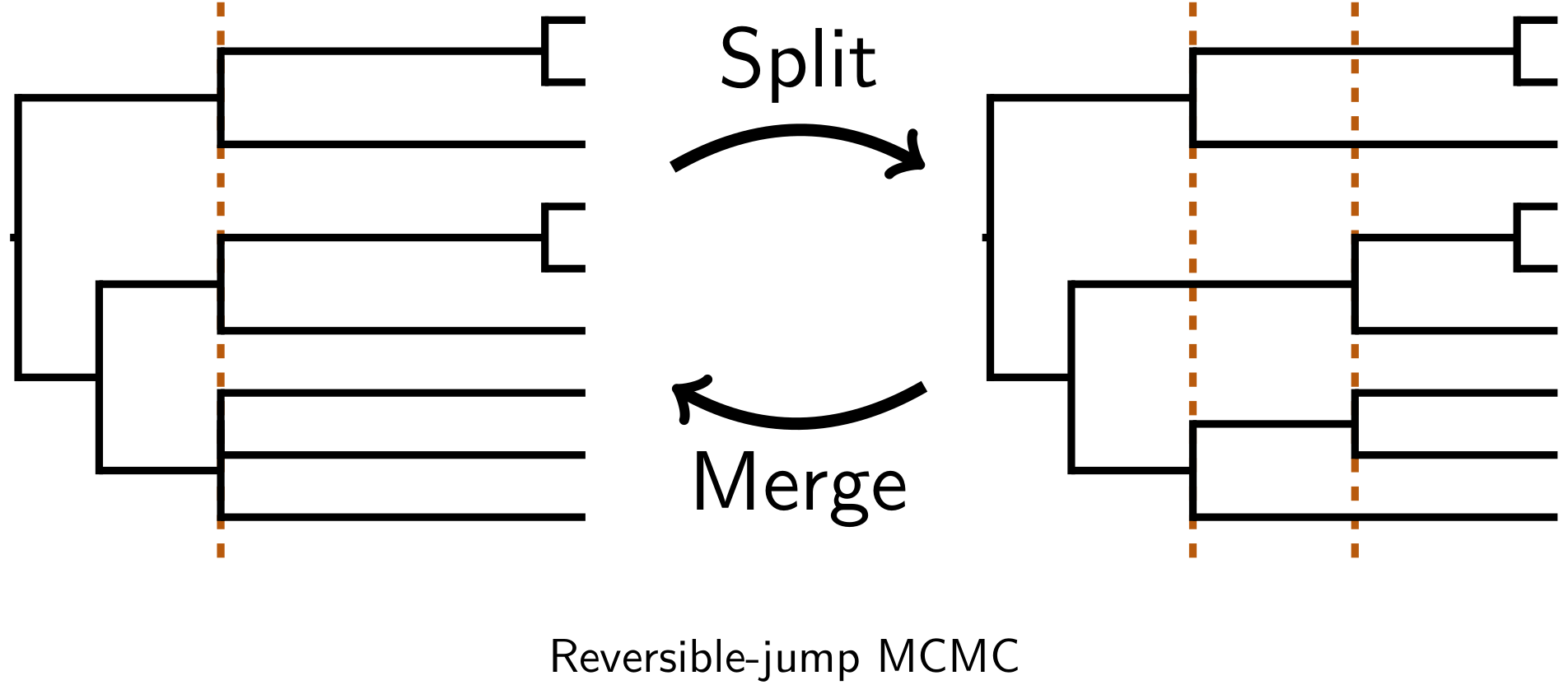
This allows us to jointly infer relationships, shared and multifurcating divergences, and divergence times.
We coupled the generalized tree model with the “SNAPP likelihood” (Bryant et al., 2012) for directly calculating the probability of biallelic characters given a population (or species) phylogeny, while analytically integrating over all possible gene trees under a coalescent model and all possible mutational histories along those gene trees under a finite-sites model of character evolution. This allows us to tackle biogeographic questions by jointly inferring a species tree and shared divergences from genomic data (Oaks et al., 2022). However, the generalized tree distribution and associated algorithms are independent of the likelihood function, and so can be used with other data types and likelihood models (i.e., lots of room for future work and applications!).
Using simulations, we found this approach consistently inferred shared and multifurcating divergences with high support with moderately sized data sets.

We simulated 100 data sets on the tree above. The histograms show the posterior probabilities of correctly inferring the shared and multifurcating divergences when analyzing all the data sets under the generalized tree model.
We applied this new approach to genomic data from two genera of geckos co-distributed across the Philippines. Fragmentations of the islands caused by interglacial rises in sea level predict shared and multifurcating divergences over the last 5 million years. Our results show support for this predicted pattern.

Shared and multifurcating divergences inferred during the evolutionary history of Gekko lizards across the Philippines. From (Oaks et al., 2022).

Shared and multifurcating divergences inferred during the evolutionary history of Cyrtodactylus lizards across the Philippines. From (Oaks et al., 2022).
If you want to learn more, here’s a talk I gave at the Evolution Meetings this summer:
And you can read our preprint here.
This work was a lot of fun, but we have only scratched the surface with the generalized tree distribution. If you are interested in this work, please reach out; there’s so much more to do!
References
- Oaks JR, Wood, Jr. PL, Siler CD, Brown RM. 2022. Generalizing Bayesian phylogenetics to infer shared evolutionary events. Proceedings of the National Academy of Sciences 119:e2121036119. DOI: 10.1073/pnas.2121036119. Link.
- Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, Roychoudhury A. 2012. Inferring Species Trees Directly from Biallelic Genetic Markers: Bypassing Gene Trees in a Full Coalescent Analysis. Molecular Biology and Evolution 29:1917–1932. DOI: 10.1093/molbev/mss086. Link.