
Research
Contents
Overview
What we do in the phyletica lab: Develop phylogenetic methods, collect genetic datasets from natural populations, and apply the former to the latter, all with the goal of ending up with a better understanding of processes of diversification. This entails a variety of day-to-day tasks, including:
- Collection-based fieldwork in fun places, such as the Southeast Asian tropics and Gobi Desert.
- Molecular lab work to collect genetic data, which is becoming ever more dominated by genomic library preparation for “next-generation” sequencing (NGS) technology.
- Developing and implementing statistical methods that allow us to test our hypotheses. This allows us to frame our work around questions that excite us, rather than framing our questions around available methods.
Below are summaries of some of our research.
Inferring shared evolutionary history
One of the major themes in our work is trying to better understand biological diversification. To do so, it is important to account for processes that can affect whole groups lineages. In other words, if you imagine multiple lineages evolving through space and time along the tree of life, there are a lot of potential processes that could cause speciation and/or extinction to essentially co-occur across multiple lineages. If we are thinking on the scale of macrobiota, this can be any of the “usual suspects” in biogeography (e.g., formation of geological features that cause vicariance of whole communities of species). If we are thinking on the scale of gene family evolution, this could be a chromosomal duplication event. If we want to learn about such processes from patterns of biological diversity, we stand to benefit from explicitly incorporating them into models of evolution.
Currently, phylogenetic models assume branching (speciation) events occur independently (given the tree) across lineages. We are working on models that relax this assumption, and allow clustering of branching events across lineages. Our first attempt at relaxing this assumption is to infer the distribution of divergence times across separate, rooted, two-taxon trees using an approximate-likelihood Bayesian framework… I.e., starting simple!
Approximate-likelihood Bayesian inference of co-diversification
The idea here is pretty simple. If, for example, we are interested in whether or not three pairs of lineages diverged due to a common mechanism, what we want to do is compare all five possible models of their divergence (depicted in the cartoons below), which range from complete dependence among their divergences (far left below) to complete independence of their divergences (far right below).
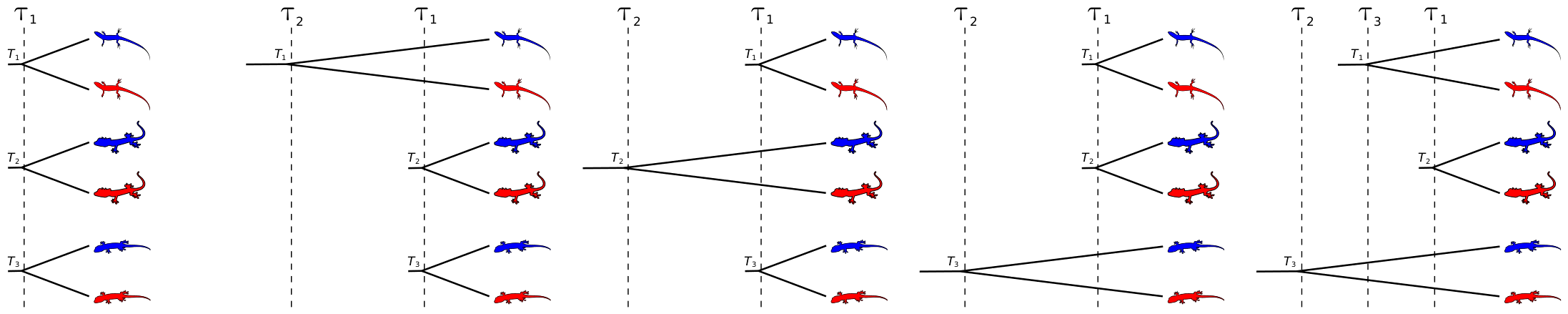
Cartoon examples of the five possible divergence models for three pairs of lineages.
The method implemented in the popular software package, msBayes, samples over these models, and uses approximate-likelihoods and a basic Monte Carlo rejection algorithm to approximate the posterior probability of each. We extended this framework, developing a new method that uses flexible prior probability distributions for many of the models’ parameters, and a nonparametric Dirichlet-process prior across all possible divergence models (Oaks, 2014). We implemented the new model in the software package dpp-msbayes, and developed a much more efficient multi-processing interface, PyMsBayes, to the models of both dpp-msbayes and msBayes.
Implementing a full-likelihood Bayesian model of co-diversification
While the method implemented in dpp-msbayes is more powerful and robust than its predecessor, msBayes, both methods are based on approximate likelihoods and discard information when reducing the genetic data to a set of insufficient summary statistics. Thus, methods struggle to discriminate among models of divergence at recent evolutionary timescales (Oaks et al., 2013; Oaks, Linkem & Sukumaran, 2014; Oaks, 2014). We are working on a method that employs a similar divergence-model-choice approach to this problem, but in a full-likelihood Bayesian framework. By utilizing all of the information in the data, we hope the method will be much more accurate and precise than its approximate-Bayesian counterparts. Ultimately, the goal is to incorporate this approach into a fully phylogenetic framework, allowing us to relax the assumption of independence of branching events across lineages and learn about shared processes of diversification via phylogenetic inference.
Diversification across Southeast Asia
One question we are actively pursuing is whether Quaternary climatic oscillations promoted diversification by fragmenting the distributions of species. An ideal model system for addressing this question is the dynamic landscape of Southeast Asia. Many groups of 26,000+ islands in this region coalesced during lower sea levels of glacial periods and were fragmented during interglacial rises in sea level.
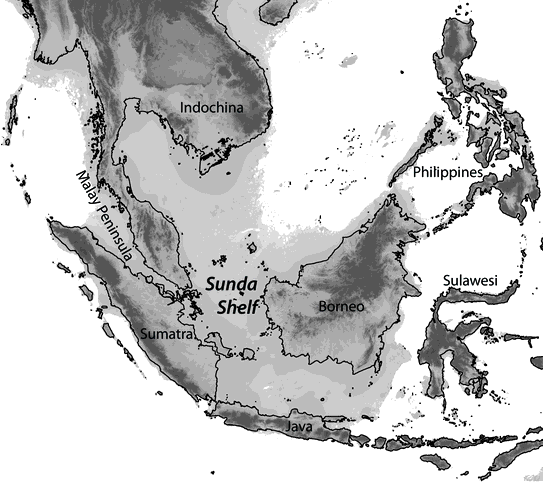
Map of Southeast Asia with land extent (depicted in gray) during glacial lowstands estimated using 120m bathymetry (data from ETOPO1).
Diversification of geckos
To test whether the repeated formation and fragmentation of island complexes promoted diversification, we are studying the evolutionary history of two genera of geckos Cyrtodactylus and Gekko that are co-distributed across most of the islands of the Philippines. So far, we have found evidence of complex histories for both genera that contradict many of the traditional areas of endemism predicted by island connectivity during glacial periods (Siler et al., 2010; Siler et al., 2012). Interestingly, for Gekko, we found evidence that the genus colonized the Philippines by “rafting” on the Palawan Microcontinental Islands that rifted away from Mainland Asia and drifted toward the rest of the Philippine Archipelago (Siler et al., 2012).
Currently, we are currently collecting genome-wide sequence data from nearly 300 individuals of Cyrtodactylus and Gekko from across the Philippine Archipelago. Combining these NGS data with new methods we are developing will allow us to better assess the affect of sea-level fluctuations on diversification across oceanic islands of Southeast Asia.
Comparative phylogeography of the Philippines
To more directly test whether glacial cycles promoted diversification, we are also taking a broadly comparative approach. For example, we are accumulating genetic data from island populations of distantly related taxa distributed across the Philippine Islands (Oaks et al., 2013). If repeated bouts of island connectivity and isolation promoted diversification, the temporal distribution of divergences across of inter-island pairs of populations should be temporally clustered and correspond to interglacial rises in sea level that fragmented the islands. Our preliminary approach to this question, using single-locus data and PyMsBayes, suggested that multiple, recent events were important in explaining the diversity across the sampled taxa (Oaks, 2014). However, due to a combination of limited data and our approximate-likelihood approach, there was too much posterior uncertainty to be confident in any divergence models. To tackle this problem, we are working with collaborators to collect more (genomic) data, and developing full-likelihood methods for inferring models of divergence that can accommodate such data.
Diversification across mainland Southeast Asia
We also want to better understand diversification in mainland Southeast Asia. Along with collaborators, we are collecting large comparative datasets of reptiles and amphibians from across Indochina and the Malay Peninsula. We are excited about applying the methods we are developing to these data to test long-standing hypotheses about the affect of biogeographical transition zones on the diversification of Sunda-Shelf biota, including the Isthmus of Kras, and current and paleo-river systems.
Diversification of the highly endemic fauna of the Gobi Desert
The Gobi is the largest desert in Asia and is a mosaic of rocky mountain ranges surrounded by basins of sand dunes. As part of an international team of collaborators (Jesse Grismer and Rafe Brown of the University of Kansas, Natalia Ananjeva of the Russian Academy of Sciences, Xianguang Guo of the Chinese Institute of Biology, and Nyamsuren Batsaikhan of the National University of Mongolia), we are working to understand the evolutionary history of the unique assemblage of reptiles that inhabit this region. Our goal is to collect genomic data from a set of focal sand and rock-adapted taxa and use the comparative methods we are developing to (1) infer their evolutionary origins, (2) test for patterns of shared evolutionary history, and (3) identify key processes that led to the diverse and unique fauna of this region.
Diversification in West-Central African rainforests
We are working with Adam Leaché and Matt Fujita to apply novel statistical methods to comparative NGS datasets to elucidate the historical processes of diversification and community assembly across West-Central African rainforests. We are particularly interested in determining how Pliocene and Pleistocene aridification cycles and associated rainforest fragmentation influenced diversification across the Afro-tropics.
Diversification of Crocodiles

Crocodyuls siamensis
How and when did crocodiles get to where they are today? How many species are there? Where and when did their common ancestor live? Motivated by these questions, and the fact that crocodiles are awesome, I collected a large dataset of DNA sequences from all extant crocodylian species and used statistical phylogenetic methods to infer the temporal and biogeographical origin of the “true” crocodiles, Crocodylus. The leading hypothesis was that Crocodylus originated in Africa, however, when was a lot more uncertain. Traditionally, crocodiles were stereotyped as an ancient group of “living fossils”, but molecular data and reassessment of paleontological data suggested the genus might be younger than previously believed.
The DNA data I collected were strongly inconsistent with the hypotheses that the common ancestor of Crocodylus was (1) ancient and (2) from Africa. Rather, my results support that all extant crocodiles shared a common ancestor from the Indo-Pacific, approximately 14–8 million years ago (Oaks, 2011). This rejects a vicariant explanation of their biogeography in favor of a recent, dispersal-mediated model involving multiple transoceanic dispersals. Interestingly, it was not until after the mid-Miocene climatic optimum, when the global climate was cooling and fellow crocodylian lineages were suffering massive extinctions, that Crocodylus radiated and dispersed around the globe. These findings raise interesting questions about how and why Crocodylus fared so well during a period with the highest crocodilian extinction rate over the last 100 million years.
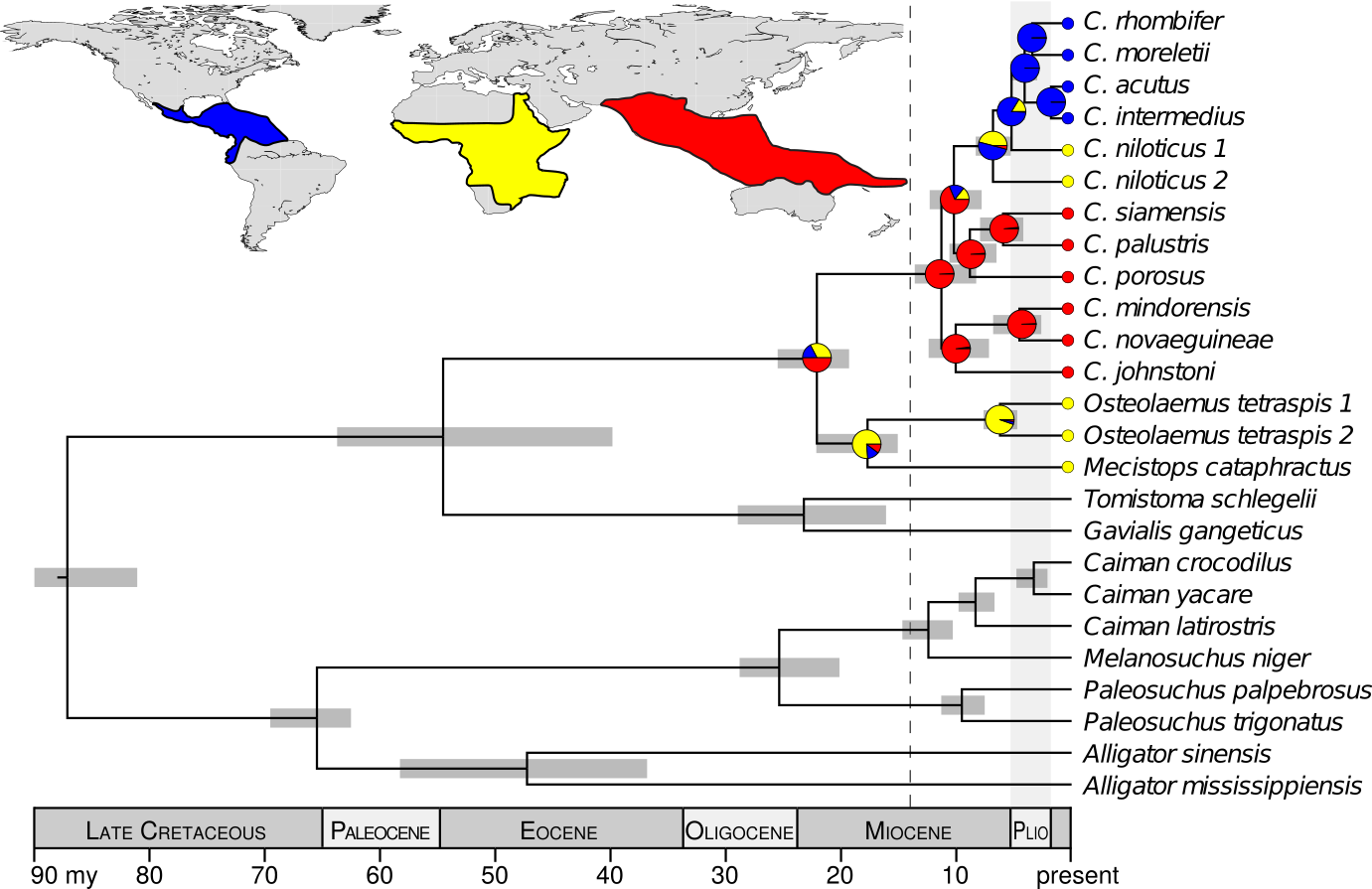
Summary of the fossil-calibrated species tree and biogeographical character-state reconstruction, supporting the Crocodylus ancestor within the Indo-Pacific and subsequent diversification after the mid-Miocene climatic optimum.
This work has been highlighted in:
Data
- We make our data available in repositories; please see our papers for specific locations. Additionally, there are some “odds and ends” available for download here.
References
- Oaks JR, Linkem CW, Sukumaran J. 2014. Implications of uniformly distributed, empirically informed priors for phylogeographical model selection: A reply to Hickerson et al. Evolution 68:3607–3617. DOI: 10.1111/evo.12523. Link.
- Oaks JR. 2014. An Improved Approximate-Bayesian Model-choice Method for Estimating Shared Evolutionary History. BMC Evolutionary Biology 14:150. DOI: 10.1186/1471-2148-14-150. Link.
- Oaks JR, Sukumaran J, Esselstyn JA, Linkem CW, Siler CD, Holder MT, Brown RM. 2013. Evidence for climate-driven diversification? A caution for interpreting ABC inferences of simultaneous historical events. Evolution 67:991–1010. DOI: 10.1111/j.1558-5646.2012.01840.x. Link.
- Siler CD, Oaks JR, Welton LJ, Linkem CW, Swab J, Diesmos AC, Brown RM. 2012. Did geckos ride the Palawan raft to the Philippines? Journal of Biogeography 39:1217–1234. DOI: 10.1111/j.1365-2699.2011.02680.x. Link.
- Oaks JR. 2011. A time-calibrated species tree of Crocodylia reveals a recent radiation of the true crocodiles. Evolution 65:3285–3297. DOI: 10.1111/j.1558-5646.2011.01373.x. Link.
- Siler CD, Oaks JR, Esselstyn JA, Diesmos AC, Brown RM. 2010. Phylogeny and biogeography of Philippine bent-toed geckos (Gekkonidae: \emphCyrtodactylus) contradict a prevailing model of Pleistocene diversification. Molecular Phylogenetics and Evolution 55:699–710. DOI: 10.1016/j.ympev.2010.01.027. Link.